

原文链接:https://pubs.acs.org/doi/10.1021/jacs.1c09535
原子催化具有原子利用率高和构效关系清晰等特点,是多相催化发展的研究重点(Nat. Chem. 2011, 3, 634−641)。分子筛作为优良的载体,规则的孔道结构赋予了其特殊的空间限域效应,利用这一结构将金属物种稳定在分子筛孔道中(M@Zeolite),可以最大程度的提高原子利用率,同时避免金属物种在催化反应中的烧结和流失。目前M@Zeolite催化剂在加氢、脱氢、氧化等反应中有着广泛的应用(J. Am. Chem. Soc. 2019, 141, 9920–9927;Angew. Chem. Int. Ed. 2020, 59, 19669–19674 )。然而贵金属封装型分子筛对配体的要求严格,且在晶化过程中容易发生沉淀、团聚等现象,因此构筑单分散的贵金属封装型分子筛仍面临着不小的挑战。
催化选择加氢是化工行业中重要的反应之一,其中,α, β-不饱和醛酮和硝基芳香化合物的选择加氢可用于制备精细化工品和高值衍生物,具有广阔的应用前景。目前工业上常用金属氢化物(NaBH4, LiAlH4)和酸性介质中的碱金属(Fe, Sn, Zn, Al)催化这两类加氢反应,但是它们都面临分离困难、产生大量固体残余等问题。Pt基催化剂自身具有优越的活化氢气的能力,因而常用于催化加氢反应中(Nature 2016, 539, 76–80;Nat. Catal. 2019, 2, 873–881),而未修饰的Pt基催化剂受限于颗粒尺寸和稳定性,无法兼顾高活性和稳定性。因此,将孤立的Pt物种稳定限域在分子筛孔道内用于α, β-不饱和醛酮和硝基芳香化合物的选择加氢是本工作的研究重点。
研究目标
1、开发出一种孤立的单位点Ptδ+物种封装型分子筛(Pt@Y);
2、利用Ptδ+…O2-这一构型实现对氢气的异裂活化,实现对α, β-不饱和醛酮以及硝基芳香化合物的高效选择加氢;
3、揭示氢气不同活化方式的成因以及选择加氢的本质。
图文精读
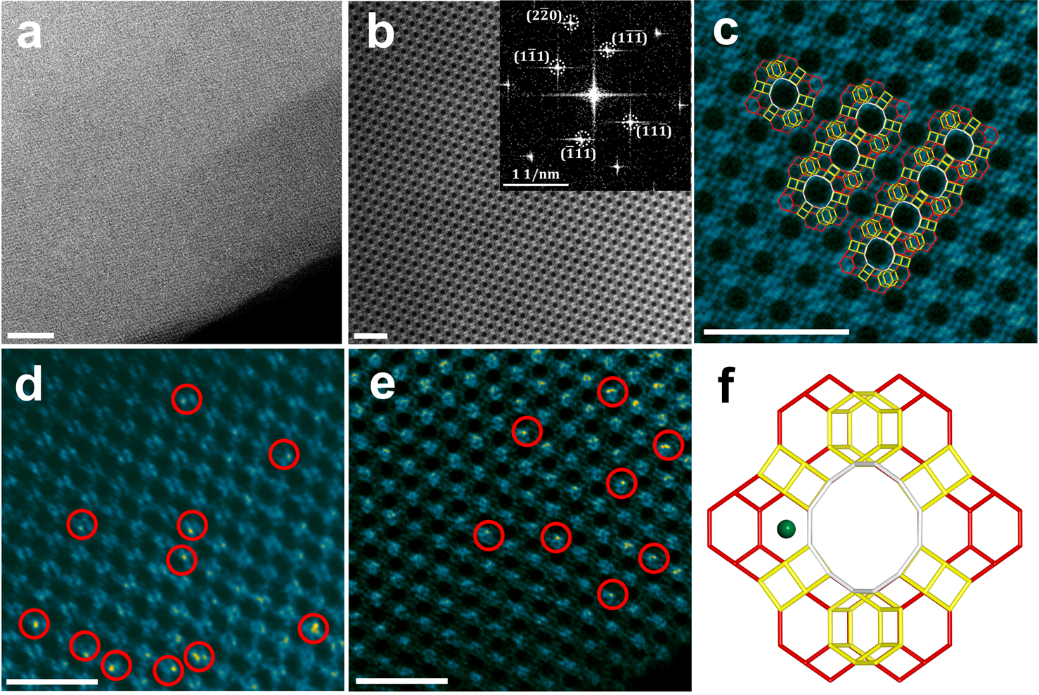
Figure 1. Electron microscopy analyses of Pt@Y (0.6 wt% Pt) zeolite. (a) Low-magnification HAADF STEM image of Pt@Y zeolite. scale bar = 20 nm; (b) Pt@Y sample viewed along the orientation of [110] and the corresponding FFT diffractogram inserted. scale bar = 5 nm; (c) Pt@Y sample viewed along the orientation of [110] as well as the schematic model on the same projection. white= supercages, red= sodalite cages, yellow= d6r cages. scale bar = 5 nm; (d, e) High-magnification Cs-corrected HAADF STEM images of Pt@Y zeolite taken with [110] incidence, indicating that only Pt single atoms are present as marked by red circles. scale bar = 5 nm; (f) The schematic model of Pt@Y along the orientation of [110]. green ball= Pt species, white= supercages, red= sodalite cages, yellow= d6r cages.

Figure 2. Microstructure characterization of Pt@Y and Pt/Y catalysts (~0.6 wt% Pt). (a) The Pt 4d XPS of Pt@Y and Pt/Y samples. (b) H2-TPR profiles of Pt@Y and Pt/Y samples (c) Pt L3-edge XANES spectra of Pt@Y, PtO2 and Pt foil; (c) FT EXAFS spectra of Pt@Y PtO2 and Pt foil; (e) FT EXAFS fitting spectrum of Pt@Y at R space; WT EXAFS spectra of PtO2 (f), Pt foil (g) and Pt@Y (h).
通过高分辨球差电镜(图1)和X射线吸收光谱(图2),可以获得Pt@Y明确的构型,即孤立的Ptδ+分散于Y型分子筛的六元环中,与骨架氧原子形成稳定的三配位构型,这一构型能够有效稳定Pt物种,避免其在反应过程中的迁移和流失。同时,Ptδ+所处的六元环被SOD笼与超笼共用,确保Ptδ+物种在后续的催化反应中与反应物分子充分接触。
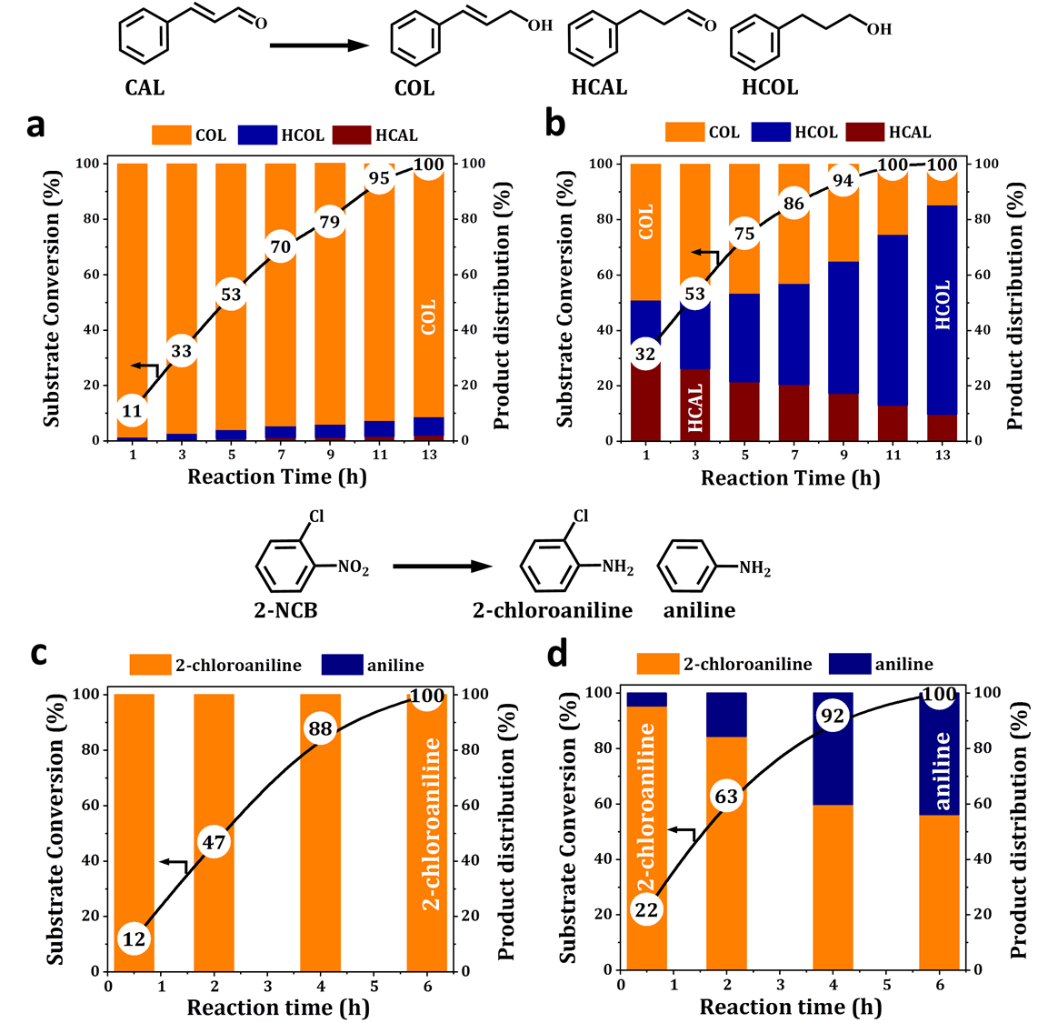
Figure 3. Selective hydrogenations over Pt@Y and Pt/Y catalysts (~0.6 wt% Pt). Hydrogenation of CAL over Pt@Y (a) and Pt/Y (b) catalysts. Reaction conditions: catalyst= 100 mg, CAL=1 mmol, isopropanol= 3 mL, temperature= 403 K, H2= 3 MPa; Hydrogenation of 2-NCB over Pt@Y (c) and Pt/Y (d) catalyst. Reaction condition: catalyst= 100 mg, 2-NCB=1 mmol, isopropanol= 3 mL, temperature= 373 K, H2= 2 MPa.

Figure 4. Dihydrogen activation on Pt@Y and Pt/Y catalysts (~0.6 wt% Pt). (a) In situ FTIR spectra of H2 activation on Pt@Y and Pt/Y recorded at 373 K; (b) In situ FTIR spectra of H2 activation on Pt@Y and Pt/Y recorded at 173 K; (c) 1H MAS NMR spectra of dehydrated Pt@Y before and after H2 adsorption. Difference spectrum obtained by subtraction of the 1H MAS NMR spectrum of dehydrated Pt@Y from that of the H2-adsorbed Pt@Y and the corresponding fitting results.
与浸渍型Pt/Y分子筛相比,Pt@Y在α, β-不饱和醛酮以及硝基芳类化合物的加氢反应中表现出优越的选择性(图3)。Pt@Y在肉桂醛全部转化的条件下对肉桂醇有92%的选择性,在邻氯硝基苯完全转化的条件下,对邻氯苯胺的选择性大于99%。为了深入探究这一高选择性的来源,本工作对氢气活化这一过程进行了原位谱学和核磁表征(图4)。通过氢气吸附原位红外,同时观测到了3714, 3668, 3565, 3466, 3245 cm-1的O-H振动峰以及2057, 1990 cm-1 Pt-H物种的信号(图4a),证明氢气在Pt@Y构型中发生了异裂,异裂后的H分别于Pt,O结合。而在氘气吸附原位红外实验中,同样观测到了2725, 2712, 2665, 2545, 2402 cm-1(O-D)以及1455, 1423 cm-1(Pt-D)的振动峰,这两种物种的偏移(νO-H/νO-D≈ 1.36,νPt-H/νPt-D≈ 1.40)进一步证明了氢气异裂活化的过程。而在氢气低温活化实验中(图 4b),浸渍型Pt/Y分子筛表现出特有的Pt-H信号(2284 cm-1),证明Pt/Y中Pt0物种促进了氢气的均裂活化(Science 2019, 363, 155−157),这一β-H物种的形成也导致了Pt/Y在反应中表现出高活性而没有选择性的加氢效果(图3)。此外,在Pt@Y氢气吸附后的1H MAS NMR谱中(图4c),也能够清楚的观测到五个O-H对应的化学位移,这一结果与原位红外的羟基振动信号可互相验证,揭示氢气异裂活化的机制。值得一提的是,在Pt/Y分子筛中也可以观测氢气异裂活化的现象,这是由于在Pt颗粒表面均裂的H原子在分子筛局域电场中发生电荷分离现象,从而产生类似异裂活化的效果。

Figure 5. Potential energy profiles of acrolein hydrogenation over Pt@Y at 0 K with optimized structures shown below.
基于Pt@Y特殊的氢气异裂活化机制以及其在催化加氢反应中高活性和选择性,本工作对Pt@Y上α, β-不饱和醛酮加氢路径进行了理论计算模拟(图5)。首先氢气在Ptδ+…O2-构型上发生异裂,生成Pt-Hδ- (hydride)和 O-H+ (proton)物种。在氢气异裂活化的基础上,分别模拟了优先吸附加氢C=O键生成丙烯醇和C=C键生成丙醛这两种路径。对于C=O双键的加氢,CH2=CHCHO*(M2)优先与Pt-H上的H结合生成CH2=CHCHOH*(M3),然后O-H上的H转移到Pt上完成二次加氢,生成CH2=CHCH2OH*(M4),这一过程需要克服0.6 eV的能垒。对于C=C双键的加氢,CH2=CHCHO*依次与PtH、O-H上的H结合,生成CH3CH2CHO*(M7),这一过程需要克服1.7 eV的能量,远高于C=O双键的加氢能垒。理论计算的结果进一步验证了Pt@Y异裂氢气的机制以及对α, β-不饱和醛酮的高选择性加氢。
本工作成功构筑了金属封装型分子筛Pt@Y,其中孤立Ptδ+存在于SOD笼与超笼共用的六元环中,与骨架氧原子配位。这一构型(Ptδ+…O2-)有助于异裂活化氢气,形成H+ and Hδ-物种,同时避免β-H物种的生成。这一氢气活化机制类似于经典路易斯酸碱对(Classical Lewis Pairs),异裂后的氢物种可用于α, β-不饱和醛酮和硝基芳香化合物的高效选择加氢。本工作为贵金属封装型分子筛的设计及其催化应用提供了一种新思路。
原文链接:https://pubs.acs.org/doi/10.1021/jacs.1c09535